 客服微v信:
客服微v信:
獨(dú)家原創(chuàng)|食欲抑制劑孕甾皂苷 P57 和果同尼皂苷 F 的研究進(jìn)展
PPS 點(diǎn)擊 藍(lán)字關(guān)注我們↑↑↑↑ 專家介紹:李 微 博士,特聘研究員,博士生導(dǎo)師,在糖化學(xué)領(lǐng)域已經(jīng)有超過(guò)十年的研究經(jīng)歷,主要從事以藥物研發(fā)為導(dǎo)向的活性糖類化合物的合成、構(gòu)效關(guān)系、靶點(diǎn)探索以及相關(guān)的化學(xué)生物學(xué)和方法學(xué)研究。2005 年畢業(yè)于武漢大學(xué)化學(xué)
專家介紹:李 微

博士,特聘研究員,博士生導(dǎo)師,在糖化學(xué)領(lǐng)域已經(jīng)有超過(guò)十年的研究經(jīng)歷,主要從事以藥物研發(fā)為導(dǎo)向的活性糖類化合物的合成、構(gòu)效關(guān)系、靶點(diǎn)探索以及相關(guān)的化學(xué)生物學(xué)和方法學(xué)研究。2005 年畢業(yè)于武漢大學(xué)化學(xué)與分子科學(xué)學(xué)院,獲得化學(xué)和生物科學(xué)雙學(xué)位。同年進(jìn)入中國(guó)科學(xué)院上海有機(jī)化學(xué)研究所,在俞飚研究員的指導(dǎo)下進(jìn)行活性甾醇和皂甙的合成研究,于 2010 年獲得博士學(xué)位,并獲得 2010年 Eli Lilly Asia Outstanding Graduate Thesis Award 一等獎(jiǎng)。之后赴美國(guó)約翰霍普金斯大學(xué)醫(yī)學(xué)院劉鈞教授課題組進(jìn)行博士后工作,主要從事 rapafucin 化合物庫(kù)的構(gòu)建和相關(guān)的化學(xué)生物學(xué)研究。2012 年年底回到上海有機(jī)所擔(dān)任副研究員,2018 年 5 月以特聘研究員職位被引進(jìn)到中國(guó)藥科大學(xué)。迄今已在 Chemical SocietyReviews、Angew Chem Int Ed 和 J Am Chem Soc 等國(guó)際重要科學(xué)期刊上發(fā)表論文多篇。
正文
食欲抑制劑孕甾皂苷 P57 和果同尼皂苷 F 的研究進(jìn)展
李國(guó)龍,李微 *
(中國(guó)藥科大學(xué)藥學(xué)院藥物化學(xué)系,江蘇 南京 211198)
[摘要]P57 和果同尼皂苷 F 是從南非火地亞仙人掌中分離得到的孕甾皂苷類化合物,具有抑制食欲的活性。主要針對(duì) P57 和果同尼皂苷 F 的化學(xué)合成、生物活性和作用機(jī)制研究進(jìn)行綜述,旨在為相關(guān)的健康產(chǎn)品和藥物研發(fā)提供參考。
肥胖是威脅人類健康的危險(xiǎn)因素之一,可引起糖尿病、心血管疾病及血脂異常等多種嚴(yán)重的疾病。進(jìn)入 21 世紀(jì)以來(lái),隨著世界上超重和肥胖人口比例的不斷攀升,肥胖的預(yù)防、控制和治療已經(jīng)成為各國(guó)醫(yī)療健康的研究重點(diǎn)。除了通過(guò)日常飲食控制和加強(qiáng)運(yùn)動(dòng)外,口服食欲抑制劑等減肥產(chǎn)品或藥物也是控制體質(zhì)量的有效途徑。火地亞(Hoodiagordonii)是一類生長(zhǎng)在非洲南部沙漠地區(qū)的仙人掌類植物,作為一種天然的食欲抑制劑被當(dāng)?shù)赝林诼L(zhǎng)的狩獵過(guò)程中食用已有上千年歷史,其功效和安全性都有所保障。實(shí)際上,火地亞提取物曾是風(fēng)靡西方國(guó)家的減肥藥,被認(rèn)為能夠用于治療超重、肥胖等相關(guān)疾病。然而火地亞的產(chǎn)量不足與成分過(guò)于復(fù)雜等因素,限制了相關(guān)產(chǎn)品開(kāi)發(fā)的后續(xù)進(jìn)展。研究表明,孕甾烷型皂苷是火地亞提取物的主要成分,而其中的孕甾皂苷 P57(1)則被認(rèn)為是抑制食欲的有效成份,作為候選藥物被研究多年。2014 年,Zhang 等報(bào)道指出,從火地亞中分離得到的果同尼皂苷(gordonoside)F(2)也是食欲抑制的活性成分。果同尼皂苷 F 與 P57 結(jié)構(gòu)類似,具有相同的苷元 hoodigogenin A(3)和相似的磁麻糖(cymarose)糖鏈,區(qū)別僅在于磁麻糖個(gè)數(shù)和末端糖基的不同。但令人感到意外的是,果同尼皂苷 F作用的靶蛋白卻與 P57 不同,提示火地亞食欲抑制活性存在著多種不同的機(jī)制。由于火地亞中的皂苷種類繁多,且在植物體內(nèi)含量過(guò)低,依靠天然提取來(lái)進(jìn)行分離純化十分困難。為了明確火地亞仙人掌中皂苷類食欲抑制成分,以求得到相應(yīng)的藥物先導(dǎo)化合物,人們對(duì) P57 和果同尼皂苷 F 進(jìn)行了相關(guān)的化學(xué)合成,以及開(kāi)展了生物活性和作用機(jī)制方面的研究。本文就上述研究進(jìn)展進(jìn)行綜述。
1 P57 的合成研究
1.1 Van Heerden 等利用氟代糖給體合成 P57
P57 的苷元 hoodigogenin A 具有少見(jiàn)的 C/D 環(huán)順式結(jié)構(gòu),在 12 位和 14 位都連有 β-OH,且 12-OH被惕格酰基所衍生化;而其糖鏈由 2 個(gè)酸敏感的高脫氧糖磁麻糖(cymarose)與 1 個(gè)黃夾竹桃糖(thevetose)通過(guò)非熱力學(xué)穩(wěn)定的 β-(1 → 4)-糖苷鍵連接而成,合成有很大難度。類似的糖鏈結(jié)構(gòu)在多種天然藥物或活性化合物中都能找到,例如地高辛、洋地黃毒苷和杠柳苷 periploside A 等。
1998 年,Van Heerden 等在一篇專利上報(bào)道了 P57 的合成研究(見(jiàn)圖 1)。他們首先通過(guò)微生物發(fā)酵的方法,利用鏈霉菌 Alonectria decora(ATCC14767)在黃體酮(4)上引入 12β-OH 和 15α-OH,得到關(guān)鍵中間體雙羥基黃體酮(5)。在經(jīng) 2 步反應(yīng)消除 15-OH 而得到 Δ14, 15 -黃體酮(6)后,他們利用乙酰氯和乙酸酐反應(yīng)體系,成功地將 3 位酮羰基烯醇化,從而構(gòu)建出 5、6 位雙鍵,隨后用乙二醇保護(hù)所得雙乙酰化底物的 20 位酮羰基,以 53% 的產(chǎn)率得到甾體 7。接下來(lái),使用硼氫化鈉還原甾體7 上的 3 位烯酯成醇,再與對(duì)甲苯磺酰氯反應(yīng)得到甾體 8。以甾體 8 為底物,利用乙酸鉀試劑構(gòu)建了三元環(huán)保護(hù)結(jié)構(gòu),接著利用四氫鋁鋰脫去乙酰基,得到二醇甾體 9。隨后,使用 N-溴代乙酰胺和吡啶將 14、15 位的雙鍵氧化成 β 構(gòu)型的環(huán)氧,然后經(jīng)四氫鋁鋰區(qū)域選擇性開(kāi)環(huán),以達(dá)到在 14 位引入 β-OH的目的。然而,在接下來(lái)用鹽酸脫除 20 位縮酮保護(hù)基的時(shí)候,Van Heerden 等發(fā)現(xiàn) 17 位乙酰基的構(gòu)型發(fā)生了異構(gòu)化,只能以約 15% 的產(chǎn)率得到目標(biāo)產(chǎn)物10。盡管甾體 10 的 14-OH 和 12-OH 的大位阻使得選擇性對(duì) 3-OH 進(jìn)行糖苷化成為可能,然而 VanHeerden 等利用氟代糖給體 11 與甾體 10 進(jìn)行氯化亞錫催化的糖苷化時(shí),卻只能以 15% 的產(chǎn)率得到所需的糖苷 12。再經(jīng)過(guò)苷元 12-OH 的惕格酰化和磁麻糖 4-O-苯甲酰基的脫除得到化合物 13 后,他們將其與二糖給體 14 進(jìn)行糖苷化,后續(xù)脫除保護(hù)基得到 P57。這些后期步驟的具體實(shí)驗(yàn)條件和相應(yīng)的產(chǎn)率在專利中并未被明確報(bào)道。
1.2 Miesch 課題組合成苷元hoodigogenin
2011 年,Miesch 課題組的 Geoffroy 等以孕甾烷衍生物 15 為原料,經(jīng) 11 步以 0.04% 的收率得到了 P57 的苷元(見(jiàn)圖 2)。首先,選擇性脫去甾體15 的 3 位乙酰基,然后經(jīng)氧化得到二酮 16。接下來(lái),使用溴/乙酸條件在 4 位引入溴后,所得溴化物在氯化鋰作用下可以轉(zhuǎn)化成 α,β-不飽和酮 17。利用與 Van Heerden 等類似的策略,Geoffroy 等在成功地將 3 位酮羰基烯醇化以構(gòu)建出 5、6 位雙鍵后,選擇用乙二醇保護(hù)所得雙乙酰底物的 20 位酮羰基,從而得到甾體 18。接著他們先利用硼氫化鈉還原得到 3αOH,然后再用氫氧化鉀水解 12 位乙酰基得到甾體二醇 19。利用 3β-OH 與 12α-OH 的位阻差異,Geoffroy等首先成功地用乙酰基選擇性地保護(hù)了 3β-OH,然后用戴斯-馬丁試劑將裸露的 12α-OH 氧化成羰基。以所得到的甾體 20 為底物,課題組借助 Norrish TypeI-Prins 反應(yīng)在 14 位引入 β-OH 而得到甾體 21,遺憾的是此關(guān)鍵步驟副產(chǎn)物較多,產(chǎn)率只有 25%。最后在 12 位引入惕格酰基并用較為溫和的碳酸鉀脫除 3位乙酰基后,即可得到苷元 hoodigogenin A。
1.3 俞飚課題組基于金催化的糖苷化策略合成 P57
2012 年,俞飚課題組的 Zhang 等利用金催化的糖基鄰己炔基苯甲酸酯給體糖苷化方法完成了P57 的化學(xué)合成。常規(guī)的糖苷化反應(yīng)多使用強(qiáng)酸性催化劑,并且伴隨著親核性副產(chǎn)物生成,導(dǎo)致反應(yīng)效率不高,底物耐受性不好。而以一價(jià)金復(fù)合物為催化劑的糖苷化反應(yīng)具有條件中性溫和、無(wú)親核物種、產(chǎn)率高、實(shí)用性廣等優(yōu)點(diǎn)。按照?qǐng)D 3 所示的逆合成分析,鑒于 P57 的糖苷鍵構(gòu)建的難度,該課題組將 P57 逆向斷裂成糖苷 22 和二糖鄰己炔基苯甲酸酯給體 23。其中糖苷 22 可進(jìn)一步逆向斷裂成苷元 3 和單糖鄰己炔基苯甲酸酯給體 24,給體22和23都可以從商業(yè)易得的原料葡萄糖轉(zhuǎn)化而得,而苷元 3 則可以由洋地黃毒苷(25)獲得。現(xiàn)就上述工作進(jìn)行詳細(xì)介紹。
1.3.1 苷元 hoodigogenin A 的合成 從 Van Heerden和 Geoffroy 等的工作中可以發(fā)現(xiàn),對(duì)苷元 14β-OH的引入存在著步驟過(guò)長(zhǎng)或者產(chǎn)率不佳等缺點(diǎn),因此Zhang 等選擇具有 12α,14β-OH 結(jié)構(gòu)的洋地黃毒苷(25)為原料來(lái)進(jìn)行苷元 hoodigogenin A 的合成。如圖 4 所示,他們依次通過(guò)選擇性乙酰化、糖鏈酸解和叔丁基二苯基硅基(TBDPS)保護(hù)反應(yīng)得到甾體 26,隨后用臭氧裂解和鋅粉/乙酸還原得到所期望的甲基酮 27。在完成 20 位羰基的保護(hù)和 3 位羥基的去保護(hù)以及氧化后,課題組利用雙(三甲基硅基)胺基鋰/三甲基氯硅烷/三乙胺條件,將底物28 的 3 位羰基烯醇化,然后通過(guò)乙酸鈀氧化成功地以 2 步 79% 的產(chǎn)率完成了 4、5 位雙鍵的引入。所得產(chǎn)物 29 可以在叔丁醇鉀存在的條件下再次發(fā)生烯醇化,促使雙鍵遷移到需要的 5、6 位,然后再借助四氫鋁鋰還原烯醇,得到具有 Δ5,6-3β-OH 結(jié)構(gòu)的產(chǎn)物 30。值得注意的是,這個(gè)過(guò)程中的烯醇中間體對(duì)氧氣極為敏感,因此反應(yīng)需要在無(wú)氧環(huán)境中才能順利進(jìn)行。課題組通過(guò) 3β-OH 的選擇性保護(hù)、惕格酸與 12β-OH 的酯化反應(yīng)得到 31,最后脫除相關(guān)保護(hù)基,合成得到了苷元 hoodigogenin A。
1.3.2 糖基鄰己炔基苯甲酸酯給體的合成 糖基給體24 的合成是以 α-D-葡萄糖甲苷(32)作為原料進(jìn)行的。經(jīng)過(guò)多步反應(yīng)得到磁麻糖甲苷(33)。隨后將 4 位羥基乙酰化,然后在 80% 乙酸、加熱的條件下,水解甲苷得到相應(yīng)的半縮醛 34,最后與鄰己炔基苯甲酸縮合成酯得到給體 24(見(jiàn)圖 5)。
在二糖給體 23 的合成方面,Zhang 等以雙丙酮-D-葡萄糖(35)作為原料。如圖 6 所示,多步反應(yīng)后得到半縮醛 36。隨后通過(guò)相應(yīng)的條件,分別制得了 Schmidt 給體(37)和鄰己炔基苯甲酸酯給體(38)。然后,他們將 Schmidt 給體與 α-D磁麻糖甲苷在三氟甲磺酸三甲基硅酯(TMSOTf)催化下進(jìn)行糖苷化反應(yīng),以 85% 的總產(chǎn)率生成二糖39。盡管此反應(yīng)是在- 40 ℃的低溫下進(jìn)行,卻能夠觀察到產(chǎn)物 39 的異頭位甲氧基有顯著的異構(gòu)化現(xiàn)象,所得到的 β-構(gòu)型比 α-構(gòu)型更多。而將鄰己炔基苯甲酸酯給體 38 應(yīng)用于此糖苷化時(shí),在室溫下用三苯基膦金(Ⅰ)三氟甲磺酸鹽(Ph3PAuOTf)進(jìn)行催化,產(chǎn)物 39 也同樣發(fā)生了異構(gòu)化。但是此條件更為溫和,所以導(dǎo)致的異構(gòu)化程度明顯小于 Schmidt給體,所得到的 α-構(gòu)型比 β-構(gòu)型更多,產(chǎn)率也有一定提升。接下來(lái),課題組將苯甲酰基替換為乙酰基,以確保在合成的最后能選擇性脫除而不影響 12 位的惕格酸酯。隨后水解甲苷 40,并與鄰己炔基苯甲酸反應(yīng)得到相應(yīng)的二糖給體 23(見(jiàn)圖 7)。
1.3.3 通過(guò)金催化糖苷化合成 P57 由于磁麻糖缺失 2位羥基,因此無(wú)法利用經(jīng)典的鄰基參與來(lái)控制 P57分子中糖苷鍵的 β-構(gòu)型。以鄰己炔基苯甲酸酯為給體的金催化糖苷化,由于在反應(yīng)過(guò)程中存在一種相對(duì)穩(wěn)定的 α-構(gòu)型中間體 41(見(jiàn)圖 8),因此有可能通過(guò)類 SN2 反應(yīng)接受受體的進(jìn)攻而表現(xiàn)出 β-選擇性。如圖 9 所示,Zhang 等以 Ph3PAuOTf 為催化劑,二氯甲烷為溶劑,發(fā)現(xiàn)在室溫下 hoodigogenin A 與鄰己炔基苯甲酸酯給體(24)的糖苷化的確表現(xiàn)出了良好的β-選擇性,所得產(chǎn)物的β-構(gòu)型/α-構(gòu)型為3.5∶1,以 58% 的產(chǎn)率得到糖苷 42。隨后用弱堿碳酸鈉有效脫除糖苷 42 非還原端的乙酰基,所得醇 22 再與二糖給體 23 在 Ph3PAuOTf 促進(jìn)下完成糖苷化。遺憾的是此處并未表現(xiàn)出明顯的 β-選擇性,所得產(chǎn)物的 β-構(gòu)型 /α-構(gòu)型為 1∶1。最后他們選擇性脫除了43β 的乙酰保護(hù)基制得糖苷 P57。
1.3.4 通過(guò)三糖糖烯合成 P57
盡管上述路線利用金催化糖苷化策略通過(guò) 20 步反應(yīng)合成了 P57,但在最后的糖苷化中并沒(méi)有表現(xiàn)出 β-選擇性,在很大程度上影響了合成效率。2018 年,Liu 等報(bào)道了利用三苯基膦氫溴酸鹽(TPHB)催化糖烯給體合成 P57的方法,在最后的糖苷化中表現(xiàn)出了令人滿意的 β選擇性(β-構(gòu)型 /α-構(gòu)型 = 6∶1)。如圖 10 所示,給體 44 上的 2 個(gè)大位阻 TBDPS 能夠有效屏蔽糖環(huán)的α 面,從而以高 β-選擇性和高產(chǎn)率完成二糖 45 的制備。通過(guò)二丁基氧化錫對(duì)二糖 45 的 4'-位平伏羥基進(jìn)行芐基保護(hù),所得化合物 46 可以通過(guò)幾步反應(yīng)轉(zhuǎn)化成磁麻二糖 47,隨后在三苯基膦金雙三氟甲磺酰亞胺(PPh3AuNTf2)催化下與黃夾竹桃糖鄰己炔基苯甲酸酯給體 38 反應(yīng),生成三糖 48。接下來(lái),該課題組使用溫和的吡啶甲酸銀試劑脫去異頭位的對(duì)甲氧基酚(MP)保護(hù)基,然后通過(guò)三氟甲磺酸酐/N,N-二異丙基乙胺條件(iPrNEt)消除異頭位羥基,得到三糖糖烯給體 49。在關(guān)鍵的糖鏈與苷元hoodigogenin A 的拼接上,他們使用 TPHB 在室溫下反應(yīng) 18 h,以 70% 的總得率和較高的立體選擇性(β-構(gòu)型 /α-構(gòu)型 = 6∶1)完成骨架拼接。最后選擇性脫除 50β 乙酰基,再次完成了 P57 的合成。
2果同尼皂苷 F 的合成研究
果同尼皂苷 F 也是從火地亞中分離出的一種四糖皂苷,與 P57 有著相同的苷元 hoodigogenin A。其糖鏈由一個(gè) D-齊墩果糖和 3 個(gè) D-磁麻糖通過(guò)β-(1 → 4)-糖苷鍵連接而成。2013 年,馬玉勇首次報(bào)道了果同尼皂苷 F 的全合成。這一合成路線效率較高,能夠以克級(jí)規(guī)模合成目標(biāo)化合物,但最后的四糖片段和 hoodigogenin A 的糖苷化立體構(gòu)型仍然難以控制。
2.1 三糖糖鏈的合成
果同尼皂苷 F 的四糖糖鏈部分可通過(guò)磁麻糖三糖片段 51(見(jiàn)圖 11)和 2- 碘代齊墩果糖給體 52(見(jiàn)圖 12)通過(guò)糖苷化拼接而成。如圖 11 所示,P57 合成路線中的中間體 45 經(jīng)由原乙酸三甲酯保護(hù)和區(qū)域選擇性開(kāi)環(huán),可以實(shí)現(xiàn)對(duì) 3 位直立羥基的選擇性乙酰化。所得化合物 53 作為受體,可以順利地與TBDPS 位阻控制的炔酸給體 44 在 Ph3PAuOTf 的催化下進(jìn)行立體專一的 β-選擇性糖苷化,以 95% 的高產(chǎn)率轉(zhuǎn)變成三糖 54。隨后馬玉勇將 TBDPS 保護(hù)基脫除得到 55,然后經(jīng)由選擇性芐基保護(hù)、甲基化和芐基脫除得到三糖受體 51。
2.2 果同尼皂苷 F 的合成
如圖 12 所示,果同尼皂苷 F 末端齊墩果糖的合成首先是以三乙酰葡萄糖糖烯(56)為原料,經(jīng)由8 步反應(yīng)制備成糖烯 57。利用 N-碘代丁二酰亞胺/乙酸條件,馬玉勇分別在糖烯的 1 位和 2 位引入乙酰氧基和碘原子,以 54% 的產(chǎn)率得到碘代物 58。然后選擇性脫去異頭位乙酰基,與三氟乙酰亞胺氯反應(yīng)得到給體 52。利用碘原子的鄰基參與作用,給體 51 與三糖片段 50 在叔丁基二甲硅基三氟甲磺酸酯(TBSOTf)作用下,以 97% 的高產(chǎn)率專一性地生成 β-構(gòu)型的四糖 59。接下來(lái)再用自由基還原脫除齊墩果糖殘基的 2 位碘原子,用吡啶甲酸銀試劑脫除異頭位的 MP 保護(hù)基,然后與鄰己炔基苯甲酸進(jìn)行縮合得到四糖給體 60。最終苷元 hoodigogenin A與四糖給體 60 的糖苷化,依然是在 Ph3PAuOTf 的催化下進(jìn)行,以高產(chǎn)率生成四糖皂苷 61(β-構(gòu)型:50%,α-構(gòu)型:48%)。遺憾的是由于缺乏有力的 β構(gòu)型導(dǎo)向性基團(tuán),此反應(yīng)幾乎沒(méi)有立體選擇性(β構(gòu)型 /α-構(gòu)型 = 1∶1)。脫除 61β 末端的乙酰基即完成了果同尼皂苷 F 的合成。
3 P57 和果同尼皂苷 F 的活性研究進(jìn)展
由于火地亞中的皂苷種類繁多,在植物體內(nèi)含量很低,分離純化十分困難,所以對(duì)其減肥活性的作用機(jī)制研究大都淺嘗輒止。其中 P57 由于被發(fā)現(xiàn)較早,其作用機(jī)制研究相對(duì)較多,但是目前依然沒(méi)有統(tǒng)一的結(jié)論。MacLean 等對(duì)大鼠下丘腦神經(jīng)元原代細(xì)胞給予 P57 后,觀察到下丘腦神經(jīng)元中 ATP含量升高 50%~150%。在對(duì)大鼠腦第三腦室給藥后,他們發(fā)現(xiàn)大鼠 24 h 內(nèi)食物攝入量下降 40% ~ 60%,從而推斷 P57 可以增加下丘腦神經(jīng)元的 ATP 含量,降低食欲,從而達(dá)到減肥的目的。Van Heerden 等對(duì)大鼠灌胃給予 P57(劑量為 6.25 ~ 50 mg · kg-1),發(fā)現(xiàn)與對(duì)照組相比,接受 P57 的大鼠其食物攝入量均明顯減少,體質(zhì)量亦減少。在與陽(yáng)性藥芬氟拉明(一種減肥藥)對(duì)比時(shí)發(fā)現(xiàn),P57 能使大鼠食欲降低從而引起體質(zhì)量減少,而芬氟拉明隨著大鼠食物攝入量的減少卻能引起大鼠體質(zhì)量的增加,表明 P57 在抑制食欲和減肥方面有著優(yōu)越性。該課題組認(rèn)為P57 抑制食欲可能和 MacLean 等所報(bào)道的通過(guò)增加下丘腦神經(jīng)元的 ATP 含量降低食欲相關(guān)。
Le Neve 等報(bào)道,在人胚胎腎 HEK 293 細(xì)胞中,P57 能夠選擇性地激活苦味受體 TAS2R7 和TAS2R14 的異源表達(dá),導(dǎo)致 HEK 293 細(xì)胞內(nèi) Ca2+濃度增加,促使膽囊收縮素(CCK)分泌,繼而通過(guò)迷走神經(jīng)傳入腸壁,產(chǎn)生飽腹感,從而發(fā)揮抑制食欲的作用。當(dāng)將 P57 的糖基或惕格酰基脫除后,則此活性喪失。因此該課題組認(rèn)為 P57 能通過(guò)激活苦味受體的表達(dá)促進(jìn) CCK 分泌,進(jìn)而發(fā)揮抑制食欲的作用。Smith 等報(bào)道,對(duì) Wistar 大鼠灌胃給予火地亞提取物(每克火地亞含有 21.3 mg 的P57),發(fā)現(xiàn)大鼠的體質(zhì)量均明顯下降,其脂肪細(xì)胞和肌纖維細(xì)胞均變小。課題組認(rèn)為 P57 是通過(guò)減少脂肪和肌肉從而達(dá)到減肥效果。而 Tsoukalas 等則報(bào)道對(duì)小腸內(nèi)分泌 STC-1 細(xì)胞給予 100 μmol· L-1的 P57 后,細(xì)胞胰高血糖素樣肽-1(GLP-1)的分泌量升高 130%。GLP-1 除了具有降糖作用之外,還可以增加飽腹感,減少食物的攝入量。因此課題組認(rèn)為 P57 可通過(guò)增加 GLP-1 的分泌來(lái)增加飽腹感,從而達(dá)到減肥目的。
GPR119 是一種與代謝密切相關(guān)的 G 蛋白偶聯(lián)受體,主要在胰島 β 細(xì)胞和胃腸道 L 細(xì)胞表達(dá)。GPR119 激動(dòng)劑通過(guò)增加細(xì)胞內(nèi)環(huán)磷酸腺苷(cAMP)的水平刺激葡萄糖依賴性的胰島素和 GLP-1 的釋放,降低血糖水平和保護(hù)胰島 β 細(xì)胞活力,能夠有效地改善患者體內(nèi)的血糖平衡。因此 GPR119 可作為靶點(diǎn)用于發(fā)現(xiàn)具有減肥活性的藥物。2014 年,Zhang 等對(duì)火地亞中的皂苷進(jìn)行篩選,發(fā)現(xiàn)只有果同尼皂苷 F 可以特異性激活 GPR119,而其他化合物包括 P57 則不能激活 GPR119。在穩(wěn)定表達(dá)GPR119 的 HEK293 細(xì)胞中,果同尼皂苷 F 能激活GPR119 介導(dǎo)的信號(hào)轉(zhuǎn)導(dǎo),表現(xiàn)為 cAMP 的積累、細(xì)胞內(nèi)鈣離子的活動(dòng)、ERK 的磷酸化與受體的脫敏和內(nèi)化。通過(guò)激活 GPR19,果同尼皂苷 F 能刺激胰島素和 GLP-1 的分泌。而在 GPR119 敲除小鼠離體胰島中,果同尼皂苷 F 則喪失了刺激胰島素分泌的能力。在小鼠的食物攝入量實(shí)驗(yàn)中發(fā)現(xiàn),當(dāng)給小鼠灌胃給予果同尼皂苷 F 200 mg · kg-1 后,小鼠 24 h食物攝入量顯著降低;而在敲除 GPR119 的小鼠中,果同尼皂苷 F 并不能降低 24 h 內(nèi)食物攝入量。因此,課題組認(rèn)為果同尼皂苷 F 能通過(guò)激活 GPR119 刺激GLP-1 和胰島素的分泌,降低食欲,進(jìn)而達(dá)到減肥的效果。另一方面,敲除 GPR119 會(huì)部分降低天然火地亞提取物的功效,說(shuō)明 GPR119 是火地亞提取物活性的重要靶點(diǎn)之一。
4 結(jié)語(yǔ)
盡管火地亞提取物曾是風(fēng)靡西方的減肥藥,但市場(chǎng)銷售的各類火地亞提取物產(chǎn)品成分復(fù)雜,P57含量很低,甚至根本不含有 P57。通過(guò)化學(xué)合成 P57及其同系物雖然不失為一個(gè)好辦法,但是該類化合物的復(fù)雜結(jié)構(gòu)在合成上具有很大的挑戰(zhàn)性。盡管目前已經(jīng)有方法來(lái)合成得到克級(jí)的化合物,但是依然存在著路線過(guò)長(zhǎng)、某些反應(yīng)不夠高效等缺點(diǎn)。特別是最后的糖苷化,由于底物缺失相關(guān)官能團(tuán)的調(diào)控,無(wú)法利用經(jīng)典的方法得到滿意的 β-選擇性,顯著降低了整體合成路線的效率。盡管利用 TBHP 促進(jìn)的三糖糖烯糖苷化可以達(dá)到不錯(cuò)的選擇性(β-構(gòu)型 /α構(gòu)型 = 6∶1),但因糖烯制備效率欠佳、穩(wěn)定性不高等缺點(diǎn),這一方法是否能夠應(yīng)用于此類化合物的大規(guī)模制備還有待確認(rèn)。此類高度酸敏感的脫氧糖β-選擇性糖苷化,依然有待開(kāi)發(fā)一些新的合成策略,例如新型離去基、添加劑、調(diào)控基團(tuán),甚至酶法合成來(lái)提升效率。
果同尼皂苷 F 能通過(guò)激活 GPR119 來(lái)抑制小鼠食欲,而 P57 則不能激活 GPR119,表明火地亞提取物是通過(guò)多種機(jī)制來(lái)共同達(dá)到顯著的食欲抑制效果,可能還存在其他的有效成分和未知的作用機(jī)制。對(duì)于這些機(jī)制的研究有希望為新型肥胖治療藥物的開(kāi)發(fā)提供更為安全有效的新靶點(diǎn)。目前有限的構(gòu)效關(guān)系數(shù)據(jù)表明,這一家族化合物的糖鏈,特別是末端糖基對(duì)化合物的活性和作用機(jī)制有著非常重要的影響。因此在后續(xù)的類似物合成和優(yōu)化研究中,可以考慮從替換末端糖基和縮減糖鏈長(zhǎng)度這 2 個(gè)方面出發(fā),同時(shí)輔以簡(jiǎn)化苷元部分(例如對(duì)合成造成很大困擾的 4、5 位雙鍵和 14 位羥基),進(jìn)行更為系統(tǒng)的構(gòu)效關(guān)系研究。
關(guān)于藥學(xué)進(jìn)展
感謝您閱讀《藥學(xué)進(jìn)展》微信平臺(tái)原創(chuàng)好文,也歡迎各位讀者轉(zhuǎn)載、引用。本文選自《藥學(xué)進(jìn)展》2020年第7期。
《藥學(xué)進(jìn)展》雜志是由中國(guó)藥科大學(xué)和中國(guó)藥學(xué)會(huì)共同主辦、國(guó)家教育部主管,月刊,80頁(yè),全彩印刷。刊物以反映藥學(xué)科研領(lǐng)域的新方法、新成果、新進(jìn)展、新趨勢(shì)為宗旨,以綜述、評(píng)述、行業(yè)發(fā)展報(bào)告為特色,以藥學(xué)學(xué)科進(jìn)展、技術(shù)進(jìn)展、新藥研發(fā)各環(huán)節(jié)技術(shù)信息為重點(diǎn),是一本專注于醫(yī)藥科技前沿與產(chǎn)業(yè)動(dòng)態(tài)的專業(yè)媒體。
《藥學(xué)進(jìn)展》注重內(nèi)容策劃、加強(qiáng)組稿約稿、深度挖掘、分析藥學(xué)信息資源、在藥學(xué)學(xué)科進(jìn)展、科研思路方法、靶點(diǎn)機(jī)制探討、新藥研發(fā)報(bào)告、臨床用藥分析、國(guó)際醫(yī)藥前沿等方面初具特色;特別是醫(yī)藥信息內(nèi)容以科學(xué)前沿與國(guó)家戰(zhàn)略需求相合,更加突出前瞻性、權(quán)威性、時(shí)效性、新穎性、系統(tǒng)性、實(shí)戰(zhàn)性。根據(jù)最新統(tǒng)計(jì)數(shù)據(jù),刊物篇均下載率連續(xù)三年蟬聯(lián)我國(guó)醫(yī)藥期刊榜首,復(fù)合影響因子0.760,具有較高的影響力
《藥學(xué)進(jìn)展》編委會(huì)由國(guó)家重大專項(xiàng)化學(xué)藥總師陳凱先院士擔(dān)任主編,編委新藥研發(fā)技術(shù)鏈政府監(jiān)管部門、高校科研院所、制藥企業(yè)、臨床醫(yī)院、CRO、由金融資本及知識(shí)產(chǎn)權(quán)相關(guān)機(jī)構(gòu)百余位極具影響力的專家組成。
《藥學(xué)進(jìn)展》編輯部官網(wǎng):www.cpupps.cn;郵箱:yxjz@163.com;電話:025-83271227。歡迎投稿、訂閱!
● 想回顧《藥學(xué)進(jìn)展》編委會(huì)主辦和協(xié)辦過(guò)的精彩活動(dòng)嗎?請(qǐng)戳這里!
→
→
→
→
→
→
→
→
→
→
點(diǎn)一下你會(huì)更好看耶
行業(yè)動(dòng)態(tài)|復(fù)宏漢霖阿達(dá)木單抗注射液HLX03通過(guò)上海市藥監(jiān)局GMP符合性現(xiàn)場(chǎng)檢查
PPS 點(diǎn)擊 藍(lán)字關(guān)注我們↑↑↑↑ 2020年9月7日,復(fù)宏漢霖(2696.HK)宣布,公司已于近日收到上海市藥品監(jiān)督管理局頒發(fā)的《藥品生產(chǎn)現(xiàn)場(chǎng)檢查結(jié)果告知書》,復(fù)宏漢霖徐匯生產(chǎn)基地順利通過(guò)上海市藥監(jiān)局針對(duì)阿達(dá)木單抗注射液HLX03的原液(DS)及制劑(DP)生產(chǎn)線的...
本文來(lái)源:藥學(xué)進(jìn)展 作者:藥學(xué)進(jìn)展 免責(zé)聲明:該文章版權(quán)歸原作者所有,僅代表作者觀點(diǎn),轉(zhuǎn)載目的在于傳遞更多信息,并不代表“醫(yī)藥行”認(rèn)同其觀點(diǎn)和對(duì)其真實(shí)性負(fù)責(zé)。如涉及作品內(nèi)容、版權(quán)和其他問(wèn)題,請(qǐng)?jiān)?0日內(nèi)與我們聯(lián)系

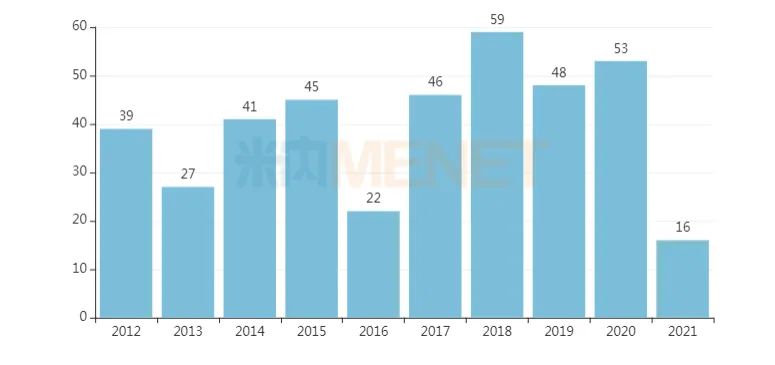
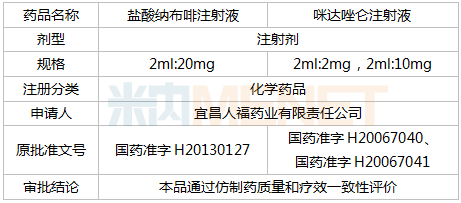
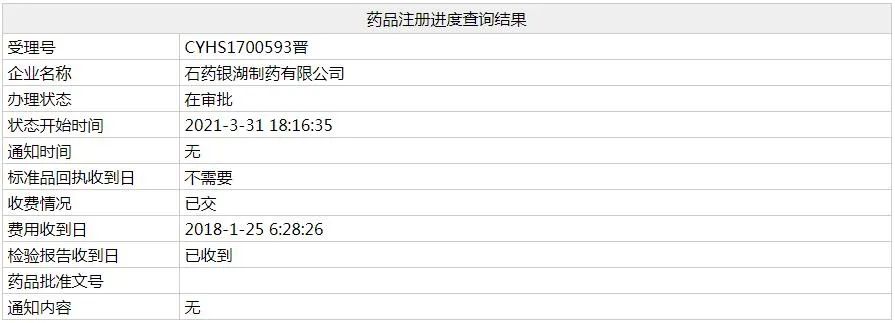
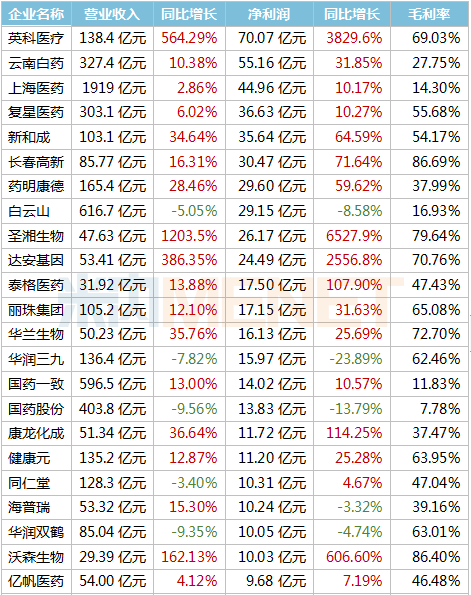





 京公網(wǎng)安備 11010802031568號(hào)
京公網(wǎng)安備 11010802031568號(hào)

