 客服微v信:
客服微v信:
8月25日,國家藥監局綜合司發布《藥品附條件批準上市申請審評審批工作程序(試行)》修訂版的征求意見稿。
藥品“附條件批準”首次正式出現在2017年的國務院文件中,此后在2020版的《藥品注冊管理辦法》中進一步細化。此次國家藥監局專門提出修改“附條件批準”,顯然是想補上此前政策中不完善的地方,進一步鼓勵藥品研發創新。
健識局注意到,新的征求意見稿中明確:某藥品獲附條件批準上市后,原則上不再同意其他同機制、同靶點、同適應癥的同類藥品開展相似的以附條件上市為目標的臨床試驗申請。
另外,附條件批準上市的藥品,在轉為常規批準之前,該品種不發布為參比制劑,其仿制藥、生物類似藥的上市注冊申請均不予受理。
這些新的提法給予了已上市的附條件批準藥品一定的市場獨占機會,這是此前中國藥品政策很少出現過的。
新藥上市,速度為王?
據國家藥監局的文件,藥品附條件上市的條件有兩種:
一種是由國家衛健主管部門等提出的公共衛生方面急需的藥品,另一種是重大突發公共衛生事件急需的疫苗。
歸納下來核心原則其實一個:公共衛生方面急需的才能夠附條件獲批上市。例如之前的新冠疫情下,口服藥、中和抗體、新冠疫苗等,基本都是通過附條件上市,以此能夠更快地應對當下突發的公共衛生狀況。
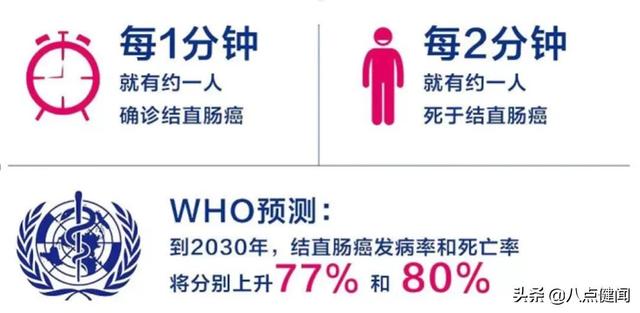
腫瘤也是人類亟待解決的重大公共衛生問題之一。近幾年,榮昌生物的維迪西妥單抗、艾力斯的甲磺酸伏美替尼、迪哲醫藥的舒沃替尼等都是附條件批準上市,及時填補臨床用藥上的空白。數據顯示,截至2022年以前,中國有48個附條件藥物產品上市,其中抗腫瘤藥物占75%左右。美國、歐盟的腫瘤藥占比情況也類似。
按照國務院辦公廳的文件:附帶條件批準上市的,只需臨床試驗早期、中期指標顯示療效并可預測其臨床價值即可。新的征求意見稿如果最終落地,搶到第一個附條件批準上市的藥品將在更長一段時間里享受獨占期。后來者要么老老實實回到常規審批通道,要么就拿出更具創新性和臨床價值的產品。

國內創新藥研發內卷是公認的事實。之前競爭最激烈的PD-(L)1,今年這股內卷之風又刮到了ADC領域,國內進入臨床階段的ADC有近60個。在眾多產品進度相近的情況下,一旦有一個通過附條件批準上市,就能率先“卡位”。同靶點、同機制、同適應癥的產品,唯有等待完成關鍵性的臨床數據才能上市。后來的仿制藥、生物類似藥更是不予受理。
這樣就給先行者賦予了擁有更大的競爭優勢,新藥上市后的競爭壓力會大大減小。
有業內分析認為,一旦有品種通過附條件批準上市,其他類似在研品種很可能會大量撤單。新的政策避免了藥物研發扎堆內卷的現象,可能對服務于藥企的CRO企業帶來一定業務壓力。
鼓勵真正有臨床價值的藥物
我國建立藥品附條件上市的制度沒有多久,新的規范調整是出于對創新的保護,而不是壓制。
國內的藥品附條件批準上市制度建立,其目的是為了鼓勵以臨床價值為導向的藥物創新,加快具有突出臨床價值的臨床急需藥品上市。速度不是唯一,臨床價值才是藥監局看重的。這次修訂從某種意義上說,是對藥品臨床價值的再強調。
附條件批準制度可以使得藥品上市速度加快,但一般來說,附條件批準上市的產品依據的是較早期的臨床試驗數據,安全性和療效還需要進一步的驗證,更加不能作為參比制劑。CDE的信號很明顯:面對急需填補的治療需求,假如臨床數據良好可以暫時補上,但總體原則一定是寧缺毋濫。
其實在同機制、同靶點、同適應癥的藥里,只允許一個附條件批準,對CDE的審評工作也是一次考驗。
健識局注意到,今年上市的武田的琥珀酸莫博賽替尼、迪哲醫藥的舒沃替尼都是附條件上市新藥,兩者的靶點和適應癥也相同,適用于EGFR 20號外顯子插入突變的局部晚期或轉移性非小細胞肺癌(NSCLC)成人患者。
如果這次新修訂的規則文件落地執行,兩個品種中有一個就暫時拿不到上市資格。
設置了市場獨占的條款之后,藥監局對附條件批準本身的審評審批也會更加謹慎,誰先誰后,可能就決定一家企業的生死。
實際上,以臨床價值為導向的趨勢,這在最近一兩年的新藥審批政策上就有所印證。2021年7月,CDE發布《以臨床價值為導向的抗腫瘤藥物臨床研發指導原則》的征求意見稿。CDE認為,當有最佳支持治療時,臨床設計應以此作為對照,而不是安慰劑。
今年7月,CDE又連發三份以患者為中心的臨床試驗設計準則,里面重點提到臨床試驗設計要以患者為中心,應避免將次優治療作為對照。
換句話說,在對照試驗中,CDE更建議企業開展頭對頭的臨床試驗。me-worse、me-too或者利用臨床設計的偽創新則不被鼓勵。鼓勵真正的臨床價值的創新,這樣的信號會越來越強烈。
新藥研發,沒有捷徑。
撰稿|楊曦霞
編輯|江蕓 賈亭
運營 | 廿十三
插圖|視覺中國
本文來源:健識局 作者:健識局 免責聲明:該文章版權歸原作者所有,僅代表作者觀點,轉載目的在于傳遞更多信息,并不代表“醫藥行”認同其觀點和對其真實性負責。如涉及作品內容、版權和其他問題,請在30日內與我們聯系










 京公網安備 11010802031568號
京公網安備 11010802031568號

