 客服微v信:
客服微v信:
變構調節(allosteric regulation)在細胞生命過程中普遍存在,包括分子間非共價相互作用、共價修飾(磷酸化、點突變,以及與小分子的化學反應)、細胞環境(溫度、輻射、pH和離子強度)和光吸收等[1–4]。變構效應精確控制著細胞的正常生命過程,包括細胞信
變構調節(allosteric regulation)在細胞生命過程中普遍存在,包括分子間非共價相互作用、共價修飾(磷酸化、點突變,以及與小分子的化學反應)、細胞環境(溫度、輻射、pH和離子強度)和光吸收等[1–4]。變構效應精確控制著細胞的正常生命過程,包括細胞信號轉導、代謝、酶的催化以及基因調控。蛋白變構調節失調與許多疾病密切相關,如癌癥、精神障礙、糖尿病、炎癥和免疫疾病等[5-7]。
變構藥物是指通過與靶蛋白上的變構位點(空間上不同于常規活性位點或底物結合位點等正構位點)相結合,從而調控蛋白生物功能的效應分子[5,8,9]。這類新型調節劑可能通過兩種方式影響蛋白的生物效應。一種是變構調節引發變構位點及其鄰近原子發生擾動并重新定向,從而產生應變能并迫使下一層原子移動,進而影響下一層原子。變構擾動通過變構波形式從變構位點傳播到正構位點,導致正構位點構象動力學微調[1,6,10]。另一種是最近提出的動力學驅動的變構概念,變構擾動遍及整個蛋白結構(包括正構位點在內),改變整個蛋白的振動模式和構象群體,可引發蛋白構象發生重大變化[11,12]。
一、變構調節劑是研發新風向
相對于傳統的正構(orthosteric)藥物,變構藥物具有幾個顯著優勢:1)克服耐藥性:在抗菌、抗病毒和抗腫瘤治療中,耐藥性問題一直是個難題,變構調節劑與正構調節劑作用于靶點的不同位點,基于不同的作用機制發揮藥效,這使變構調節劑有可能克服正構調節劑在治療過程中產生的獲得性耐藥[13];2)高選擇性:變構位點的同源序列較低,氨基酸殘基保守性低,因此具有更高的選擇性、更低的脫靶毒性和更低的給藥劑量[14];3)解決難成藥靶點困境:有些正構位點口袋殘基高度保守,且為極性和帶電荷環境,不僅導致調節劑缺乏選擇性、毒性高,而且導致調節劑的細胞滲透性和口服生物利用度差而缺乏成藥性;其次,正構位點的界面大且平坦,缺少合適的小分子結合口袋(如蛋白相互作用界面類靶點);又或者天然底物(ATP/GTP)在細胞中的濃度非常高,且與活性位點的親和力非常強,使得直接靶向正構位點的調節劑極難發揮作用等[15-16];4)變構調控可能更精準:有些蛋白功能窗口較小,既需要調控又不能過度調控,而變構調節劑可以雙向調節蛋白功能,發揮精準調控作用,如變構激動劑更不容易誘導蛋白受到激活作用后脫敏[17]。
由于變構調節藥物顯示出巨大的開發前景及其較傳統正構藥物的顯著優勢,靶點變構效應已被確立為藥物發現的新機制,引發了變構藥物發現、優化以及臨床開發的熱潮,吸引了眾多生物科技公司和大型制藥企業涌入這一熱門領域,觸發了許多大額融資、合作與交易[18]。以下,我們將以BCR-ABL1變構抑制劑為例,全面介紹這類新型藥物的發展歷史、作用機制、臨床進展、應用潛力等多方面內容。
二、BCR-ABL1抑制劑發展歷史
伊馬替尼作為首個上市的靶向治療藥物,開創了小分子靶向治療腫瘤的時代,被美國《時代》周刊譽為”銀色子彈“。這款BCR-ABL1抑制劑的成功研發使得原來致死率很高的慢性粒細胞白血病(CML)變成了慢性病,CML的治療也因此獲得了里程碑式的進步[19]。然而,10%~20%的CML患者在接受伊馬替尼治療后產生了獲得性耐藥,主要原因是BCR-ABL1基因可產生50多種點突變[20]。雖然后續幾種二代藥物的上市使CML的治療方案得以革新,為伊馬替尼耐藥的CML患者帶來了更多的臨床獲益,但是大多數患者仍須持續用藥,而且最棘手且最常見的耐藥突變BCR-ABL1T315I對目前所有一、二代BCR-ABL1抑制劑均耐藥。盡管在國外上市的泊那替尼(Ponatinib)可以克服BCR-ABL1T315I耐藥,但該藥會引發與治療相關的動脈血栓形成和肝臟毒性風險,并且體外實驗顯示,一些少見的單點突變以及多重突變(compound mutations)也會對泊那替尼產生耐藥[21]。目前,仍有多家國內外藥企在積極開發第三代BCR-ABL1抑制劑,但它們依然是通過與ATP競爭活性位點的正構抑制劑,且結構與泊那替尼相似,因此與泊那替尼類似的安全性以及交叉耐藥(cross resistance)問題依然存在。
為克服上述BCR-ABL1T315I以及對泊那替尼單點或多重突變耐藥、解決現有藥物的不耐受性和毒性問題,制藥巨頭諾華率先布局了第四代BCR-ABL1變構抑制劑(ABL001),目前該項目正在開展III期臨床研究;國內方面,進展最快的是深圳市塔吉瑞生物醫藥有限公司(以下簡稱“塔吉瑞”)的TGRX-678,該BCR-ABL1變構抑制劑已經進入臨床開發階段。
三、BCR-ABL1變構抑制劑的作用機制
通常情況下,ABL1激酶域的變構位點被其自身N端肉豆蔻酰肽占據,誘導SH3-SH2-Kinase結構域發生交聯,可發揮負向調控ABL1激酶活性的關鍵作用,調控著細胞正常增殖信號的傳導(圖1)。當BCR與ABL1基因融合后,BCR-ABL1激酶的N端肉豆蔻酰肽丟失,導致ABL1激酶域上的變構位點空缺,無法誘導SH3-SH2-Kinase結構域交聯,自抑制平衡被打破,處于活化開放構象,致使BCR-ABL1激酶持續激活,誘發細胞增殖和腫瘤形成(圖2)[22,23]。
圖1. ABL1激酶的N端肉豆蔻酰肽負向調控示意圖

圖2. BCR-ABL1融合激酶持續激活調控示意圖
為了恢復ABL1激酶負向調控功能,諾華和塔吉瑞通過模擬天然ABL1激酶肉豆蔻酰N端肽的功能,分別開發了ABL001 和TGRX-678。這兩個分子與激酶結構域C端肉豆蔻酰空缺變構位點結合(圖3a紫色球形區域),使得SH3和SH2結構域交聯到激酶結構域,將ABL1激酶由活性致癌狀態轉變為“封閉”失活非致癌狀態,重新穩定了ABL1激酶的自抑制構象(圖3),從而發揮抗腫瘤作用[24]。
圖3.TGRX-678與ABL1激酶別構位點結合誘導SH3、SH2結構域與Kinase結構域交聯,使ABL1激酶由活性致癌狀態3a轉變為失活非致癌狀態3b
由此可見,第四代BCR-ABL1抑制劑的作用位點和機制與前三代正構抑制劑截然不同。第一代抑制劑伊馬替尼、第二代抑制劑達沙替尼、尼洛替尼、博蘇替尼、拉多替尼、氟馬替尼,以及第三代抑制劑泊那替尼的分子機制是與ATP結合位點“鉸鏈區”結合(圖3a紅色球形區域),為ATP競爭性抑制劑,抑制BCR-ABL1激酶的自身磷酸化和底物磷酸化,從而抑制癌細胞的增殖和腫瘤形成,均為典型的正構抑制劑。ABL001和TGRX-678 作用于BCR-ABL1激酶域的變構位點,其位于ABL1激酶催化結構域的C端肉豆蔻酰結合位點(圖3a紫色球形區域),遠離正構ATP活性位點及T315I突變氨基酸殘基,因此ABL001和TGRX-678 與BCR-ABL1結合幾乎不受T315I的影響。
ABL001和TGRX-678 的變構調節作用不僅能夠通過變構“波”的形式引發正構位點構象動力學微調,使得對T315I耐藥無效的尼洛替尼重新與BCR-ABL1T315I激酶正構位點結合,恢復其抑制作用。此外,變構抑制劑動力學驅動的變構效應,導致SH3和SH2結構域交聯到激酶結構域,使整個蛋白發生重大構象變化,從而形成失活非致癌狀態。盡管BCR-ABL1Y253H/T315I多重突變對泊那替尼耐藥,但有研究提出泊那替尼與ABL001聯用時,泊那替尼可短暫性占據正構活性位點,誘導構象從DFG-in活性構象轉變為DFG-out非活性構象,這種構象的改變大大降低了肉豆蔻酰變構位點的柔性,使其能夠組裝并與ABL001更好的結合。反過來,ABL001與變構位點結合后預計將導致泊那替尼與BCR-ABL1激酶的DFG-out非活性構象進一步持續、穩定的結合,這種”正構-變構“相互協同的調節作用機制,最終導致強有力的激酶抑制,以克服對泊那替尼的突變耐藥性[21]。因此,正構抑制劑與變構抑制劑聯合可發揮強大的協同作用,產生意想不到的療效。
四、BCR-ABL抑制劑臨床前及臨床試驗結果
臨床前研究表明,0.1nM級別以上的ABL001無論在ATP低濃度(10uM)還是高濃度(2mM) 條件下,對激酶的抑制率相當,表明ABL001是有效的BCR-ABL1非ATP競爭性激酶抑制劑。ABL001對BCR-ABL1野生型、T315I及其它突變型的Ba/F3細胞都有較強的生物活性(圖4),可有效針對正構抑制劑引起的獲得耐藥性問題,特別是第一、二代無法克服的守門殘基T315I突變耐藥[24]。
圖4. 變構抑制劑與正構抑制劑對BCR-ABL1及其各種突變型的Ba/F3細胞系的活性
此外,多種實驗對ABL001的選擇性進行了評估。在ABL001濃度<10uM下,不會抑制其它60多種重組激酶的磷酸化作用[24]。采用450種腫瘤細胞,對ABL001變構抑制劑和正構抑制的活性和選擇性進行評估發現,ABL001對BCR-ABL1陽性腫瘤細胞表現出優異的抗增殖活性(IC50=1-20nM),而對BCR-ABL1陰性腫瘤細胞的抑制活性較低(IC50>1000nM),并且其選擇性優于正構抑制劑,如圖5所示。此外,濃度<3uM的ABL001對G蛋白偶聯受體、細胞轉運蛋白、離子通道、核受體和酶無實質性影響[25]。ABL001不僅克服第一、二代抑制劑的耐藥性,還凸顯出其高度的選擇性,可能避免出現第三代抑制劑泊那替尼引起的嚴重毒副作用。臨床前研究還表明,ABL001與尼洛替尼聯用,這一互補組合達到了很強的治療效果,可完全控制異種移植腫瘤小鼠的腫瘤進展,且停止給藥后不易復發(圖6)[24]。ABL001與泊那替尼聯用,不僅對泊那替尼最強耐藥的多重突變Y253H/T315I、E255V/T315I等產生活性,還能有效抑制BCR-ABL1多重突變的出現并逆轉泊那替尼的耐藥,系統地解決了因多重突變造成的耐藥性問題,且有望大大降低臨床所需泊那替尼的濃度,減少副作用,為CML患者提供一種有效的治療策略[21]。
圖5. BCR-ABL1抑制劑在BCR-ABL陽性和陰性腫瘤細胞的活性和選擇性
圖6. ABL001與尼洛替尼聯用治療KCL-22細胞的小鼠移植瘤模型的效果
臨床研究表明,在對既往正構抑制劑耐藥或發生不可接受的副作用,并且接受過至少兩種正構激酶抑制劑治療的CML患者,包括泊那替尼治療失敗的患者和有T315I突變的患者中,ABL001表現出較好的安全性和有效性[26]。ABL001與伊馬替尼聯合應用的臨床試驗表現出有希望的初步療效,且具有良好的安全性和耐受性[27]。ABL001的一項III期臨床試驗ASCEMBL結果顯示其與博蘇替尼(bosutinib)相比,24周的主要分子學緩解(MMR)率顯著且更優,研究達到主要終點[28]。研究人員正在評估ABL001與其它正構激酶抑制劑聯合用作一線CML治療方案。ABL001在治療CML的臨床試驗中已初顯成效,預示著顯著的臨床藥用價值。
TGRX-678是國內首個且研發進展最快的第四代BCR-ABL1變構抑制劑,也是全球第二款BCR-ABL1變構抑制劑,目前已獲得中國臨床許可(受理號:CXHL2000158/CXHL2000159),正在積極籌備臨床試驗。臨床前體內外研究結果顯示,與ABL001相比,TGRX-678對Ba/F3 Bcr-AblT315I細胞的活性和選擇性更高,口服生物利用度更佳,動物體內安全性也優于ABL001,有潛力成為best-in-class,因此其在臨床上的表現令人期待。
五、總結與展望
基于獨特的作用位點和機制,BCR-ABL1變構抑制劑不僅可以用于治療既往對正構抑制劑耐藥(包括守門殘基T315I突變耐藥)或不耐受的CML患者,與正構抑制劑的聯合應用還能夠克服泊那替尼單點和多重耐藥,降低耐藥性的發生,逆轉藥物的獲得耐藥性,減少給藥劑量,降低毒副作用,有望使患者長期維持無治療緩解,實現功能性治愈。BCR-ABL1變構抑制劑還有望用于治療其它腫瘤或疾病,大幅拓展其臨床適應癥和藥用價值。期待這類新型藥物能夠開啟靶向治療的新篇章,為更多克服耐藥性藥物的研發指引新方向。
參考文獻:
[1] Nussinov, R.and Tsai, C.J. (2013) Allostery in disease and in drug discovery. Cell 153,293–305.
[2] Lu, S. et al.(2014) Harnessing allostery: a novel approach to drug discovery. Med. Res. Rev.34, 1242–1285.
[3] Fenton, A.W.(2008) Allostery: an illustrated definition for the ‘second secret of life’.Trends Biochem. Sci. 33, 420–425.
[4] Nussinov, R.et al. (2014) Principles of allosteric interactions in cell signaling. J. Am.Chem. Soc. 136, 17692–17701.
[5] Tsai, C.J.and Nussinov, R. (2014) A unified view of ‘how allostery works’. PLoS Comput.Biol. 10 e1003394.
[6] Goodey, N.M.and Benkovic, S.J. (2008) Allosteric regulation and catalysis emerge via acommon route. Nat. Chem. Biol. 4, 474–482.
[7] Liu, J. and Nussinov, R. (2016) Allostery: an overview of its history, concepts, methods,and applications. PLoS Comput. Biol. 12 e1004966.
[8] Changeux,J.P. and Christopoulos, A. (2017) Allosteric modulation as a unifying mechanismfor receptor function and regulation. Cell 19, 4–21.
[9] Lu, S. et al.(2019) Allosteric methods and their applications: facilitating the discovery of allosteric drugs and the investigation of allosteric mechanisms. Acc. Chem.Res. 52, 492–500.
[10] Berezovsky,I.N. et al. (2017) Protein function machinery: from basic structural units to modulation of activity. Curr. Opin. Struct. Biol. 42, 67–74.
[11] Ahuja, L.G.et al. (2019) Tuning the ‘violin’ of protein kinases: the role of dynamicsbased allostery. IUBMB Life 71, 685–696.
[12] Kornev, A.P.and Taylor, S.S. (2015) Dynamics-driven allostery in protein kinases. Trends Biochem. Sci. 40, 628–647.
[13] Duan, Ni& Li, Yun & Yuran, Qiu & Pu, Jun & Lu, Shaoyong & Zhang,Jian. (2020). Combining Allosteric and Orthosteric Drugs to Overcome Drug Resistance. Trends in Pharmacological Sciences. 41.
[14] Grover A, K.Use of Allosteric Targets in the Discovery of Safer Drugs. Med Princ Pract2013;22:418-426.
[15] Arkin,Michelle. Finding Small Molecule Ligands for Protein-Protein Interactions and Other“Undruggable”Targets.Biophysical Journal, Volume 98, Issue 3, 411a.
[16] Kessler D,Gmachl M, Mantoulidis A, et al. Drugging an undruggable pocket on KRAS. Proc Natl Acad Sci U S A. 2019;116(32):15823-15829.
[17] Gjoni T,Urwyler S. Receptor activation involving positive allosteric modulation, unlikefull agonism, does not result in GABAB receptor desensitization. Neuropharmacology.2008;55(8):1293-1299.
[18] Jarvis LM.Drug hunters explore allostery’s advantages. Chemical & Engineering News.2019 March 10.
[19] Henderson CAJr. Imatinib: the promise of a "magic bullet" for cancer fulfilled. J Med Assoc Ga. 2003;92(1):12-22.
[20] Apperley J.Part I: Mechanisms of resistance to imatinib in chronic myeloid leukaemia.Lancet Oncol 8:1018,2007-1029.
[21] Eide CA,Zabriskie MS, Savage Stevens SL, et al. Combining the Allosteric Inhibitor Asciminib with Ponatinib Suppresses Emergence of and Restores Efficacy against Highly Resistant BCR-ABL1 Mutants. Cancer Cell. 2019;36(4):431-443.e5.
[22] Hantschel O,Superti-Furga G. Regulation of the c-Abl and Bcr-Abl tyrosine kinases. Nat Rev Mol Cell Biol. 2004;5(1):33-44.
[23] Adrián FJ,Ding Q, Sim T, et al. Allosteric inhibitors of Bcr-abl-dependent cell proliferation. Nat Chem Biol. 2006;2(2):95-102.
[24] Wylie, A.,Schoepfer, J., Jahnke, W. et al. The allosteric inhibitor ABL001 enables dual targeting of BCR–ABL1. Nature 543, 733–737 (2017).
[25] Schoepfer J,et al. Discovery of Asciminib (ABL001), an Allosteric Inhibitor of the Tyrosine Kinase Activity of BCR-ABL1. J Med Chem. 2018;61(18):8120-8135.
[26] Hughes TP,et al. Asciminib in Chronic Myeloid Leukemia after ABL Kinase Inhibitor Failure. N Engl J Med. 2019; 381(24):2315-2326.
[27] Cortes J, etal. Combination Therapy Using Asciminib Plus Imatinib (IMA) in Patients (PTS)with Chronic Myeloid Leukemia (CML): Results from a Phase 1 Study. Poster presented at: EHA Annual Meeting; June 15, 2019.
[28] Novartis investigational novel STAMP inhibitor asciminib (ABL001) meets primary endpointof Phase III chronic myeloid leukemia study. News Release. Novartis. August 26,2020. Accessed August 26, 2020. https://bit.ly/2Qpmc7c
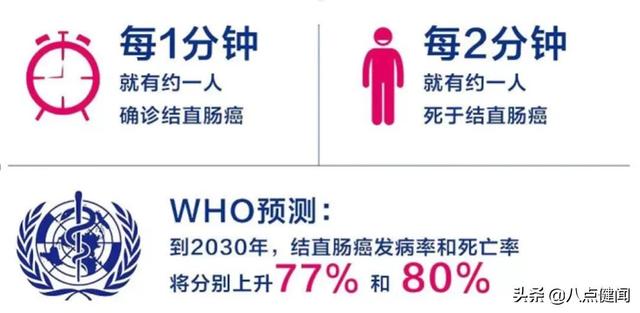
只需注射1針!強生新冠疫苗啟動III期臨床,預招6萬名志愿者
在激烈的新冠疫苗競賽中,多款候選產品已進入最后試驗階段。國內方面,目前已有11款新冠疫苗進入臨床試驗階段,其中4款進入III期臨床。9月22日,由中國工程院院士陳薇團隊和康希諾生物合作研發的重組新冠疫苗(Ad5-nCoV)國際Ⅲ期臨床試驗在巴基斯坦取得初步...
本文來源:醫藥魔方Plus 作者:小編 免責聲明:該文章版權歸原作者所有,僅代表作者觀點,轉載目的在于傳遞更多信息,并不代表“醫藥行”認同其觀點和對其真實性負責。如涉及作品內容、版權和其他問題,請在30日內與我們聯系










 京公網安備 11010802031568號
京公網安備 11010802031568號

