 客服微v信:
客服微v信:
編譯丨柯柯 BioMarin Pharmaceutical日前宣布,美國FDA對其基因療法valoctocogene roxaparvovec用于重度A型血友病的生物制品許可證申請(BLA)發出了一份完整回復函(CRL),以表明申請的審查周期已經完成,但該申請并沒有獲得批準。受此消息影響,BioMarin股
BioMarin Pharmaceutical日前宣布,美國FDA對其基因療法valoctocogene roxaparvovec用于重度A型血友病的生物制品許可證申請(BLA)發出了一份完整回復函(CRL),以表明申請的審查周期已經完成,但該申請并沒有獲得批準。受此消息影響,BioMarin股價于19日開市前下跌超20%。目前,該療法仍在歐洲藥品管理局接受審查。

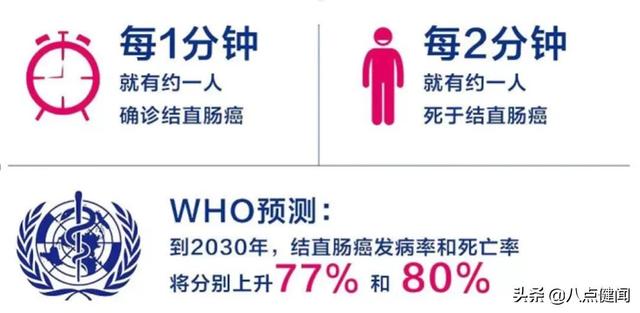
據悉,該申請基于3期臨床研究的中期分析數據,以及來自1/2期研究的三年數據。3期臨床試驗結果顯示,16例接受治療的患者平均年出血率(ABR)為1.5,與接受標準預防療法的患者相比降低了85%;同時,年度凝血因子VIII用量減少84%,在接受治療后5-26周之間,患者的年度凝血因子VIII用量減少94%。三年長期隨訪結果表明,患者的平均ABR為0.7,中位ABR為0;年度凝血因子VIII用量在第三年與對照組相比減少了96%。此前,FDA就上述BLA所需數據的范圍達成了一致意見。但在CRL中,FDA對BioMarin正在進行的3期臨床研究270-301中的兩年數據提出了新的建議,要求以ABR為主要終點,提供持久效果的實質證據。BioMarin表示,在開發或審查期間的任何時候FDA都沒有提出這一建議。同時,FDA建議BioMarin完成3期臨床研究,并提交所有研究參與者的兩年隨訪安全性和有效性數據。FDA的結論是,1/2期臨床研究270-201和3期臨床研究之間的差異,限制了依靠1/2期研究來支持該基因療法療效持久性的能力。3期研究于2019年11月全面注冊,最后一例患者將于2021年11月完成兩年隨訪。雖然BioMarin聲稱FDA首次提出這一新建議,但也說明,FDA擔心的是該基因療法的持久性。2019年5月報告的三年數據顯示,患者在接受治療12-18個月后體內的凝血因子VIII水平有所下降,這引發了人們對該療法持久性的擔憂,增加了患者可能需要重新給藥以防止出血的可能性。對于這次“拒絕”,BioMarin計劃在未來幾周與該機構會面,就下一步獲得批準達成一致。A型血友病,也稱為凝血因子VIII缺乏癥或經典血友病,是一種由凝血蛋白凝血因子VIII缺失或缺陷引起的X連鎖遺傳病,大約每10000人中就有1人患該病。盡管該疾病主要由遺傳導致,但約1/3的病例是由自發突變引起的。重度A型血友病個體約占該患病人群的50%,其護理標準是預防性方案:靜脈注射替代因子VIII,每周最多2-3次(每年100-150次)。盡管有這種治療方法,許多人仍然會經歷突發出血,導致逐漸衰弱的關節損傷,對他們的生活質量產生重大影響。Valoctocogene roxaparvovec是一種使用AAV5載體遞送表達凝血因子VIII的轉基因的基因療法,其優勢在于可能只需要一次治療就能獲得表達凝血因子VIII的基因,從而不再需要長期接受預防性凝血因子注射。基于這一優勢,該基因療法上市后很可能會讓目前的A型血友病產品“如臨大敵”,且首先與羅氏的抗體藥物Hemliba展開競爭。今年對60名美國血液學家的調查顯示,若valoctocogene roxaparvovec順利獲批,預計在一年內將有29%的Hemliba患者和34%的凝血因子VIII患者轉為基因治療。參考來源:BioMarin Receives Complete Response Letter (CRL) from FDA for Valoctocogene Roxaparvovec Gene Therapy for Severe Hemophilia A
END
我知道你在看喲
本文來源:新浪醫藥 作者:柯柯 免責聲明:該文章版權歸原作者所有,僅代表作者觀點,轉載目的在于傳遞更多信息,并不代表“醫藥行”認同其觀點和對其真實性負責。如涉及作品內容、版權和其他問題,請在30日內與我們聯系










 京公網安備 11010802031568號
京公網安備 11010802031568號

